- Are you researching human diseases?
- Do you need to interpret a large human DNA sequencing dataset?
- Is your time and effort valuable?
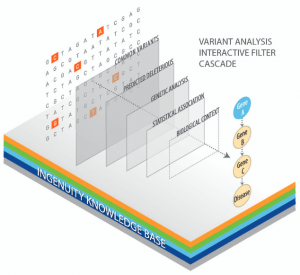
Introducing Ingenuity Variant Analysis (IVA)—IVA helps biomedical researchers quickly discover disease-causing variants, with minimal false leads. It is a HIPAA-certified web platform for comparing and annotating comprehensively sequenced human genomes to shortlist compelling gene variants via an interactive series of filters. This is critical for the identification of variants most likely to impact symptoms, biological processes, signaling pathways, and/or genes known to be implicated in disease progression or drug response.
IVA leverages the information contained in the expert-curated Ingenuity Knowledge Base, which contains over four million gene and variant findings described in the literature and public databases such as OMIM, TCGA, and COSMIC, to analyze hundreds of samples in just hours. It is a gateway to thousands of high quality, phenotypically and ethnically diverse human genome samples that serve as healthy controls for rapid discovery and validation of causal elements of disease.
During the IVA workflow, all gene variants within a dataset are compiled and then narrowed down through an interactive series of filters that can be adapted to reflect phenotypes and biologically relevant criteria of interest. Filters can be selected for consistency with Mendelian inheritance patterns, the use of single and bidirectional statistical burden tests to compare deleterious variants between study groups, and quality considerations, such as call confidence and PASS status from upstream variant calling pipeline and read depth.
The newest release of IVA includes a Phenotype-Driven Ranking filter that computes a score representing the compatibility between phenotype profile and disease to rank variants that reside in disease-implicated genes and a filter that takes into account variant frequency in ExAC ethnic subpopulations.
IVA is easy to use and requires no specialized bioinformatics skills or additional resources. Register for IVA with your Pitt e-mail address. HSLS Molecular Biology Information Service (MBIS) will teach a hands-on workshop on Variant Detection & Analysis on March 29; please register to attend. Contact MBIS with any additional IVA questions.
~Carrie Iwema