Researchers have been finding variants and mutations using Sequencher software for a long time. Sequencher’s powerful algorithms and ease of use have made it a staple for genetic researchers all over the world. One of the earliest studies linking the Human Mutator Gene Homolog MSH2 to colon cancer featured Sequencher as the software of choice.
The feature list that Sequencher offers for analyzing Sanger data is robust. You’ll be amazed at the plethora of tools available, including proprietary algorithms you won’t find anywhere else. Sequencher has three different alignment algorithms for assembling Sanger data. Each one is tuned for varying levels of sequence quality and genetic variation. You also have the option to adjust the parameters for each algorithm and save them as a template, saving you time on the next project. Sequencher gives you several options for how the resulting consensus bases are called. Choices range from a simple majority rule to a rigorous forensic standard.
Once the alignment is complete, you’ll have access to one of Sequencher’s most powerful features, the Variance Table. This table empowers you to quickly and accurately find all of the variants in your data. Using the Review Mode you can simultaneously view the Variance Table, the contig alignment, the corresponding chromatogram traces, and the Translated Variance Table. All four panes are linked: clicking on a base in one will highlight your selection in all four. Below is a screenshot showing an MSH2 analysis in the Variance Table.
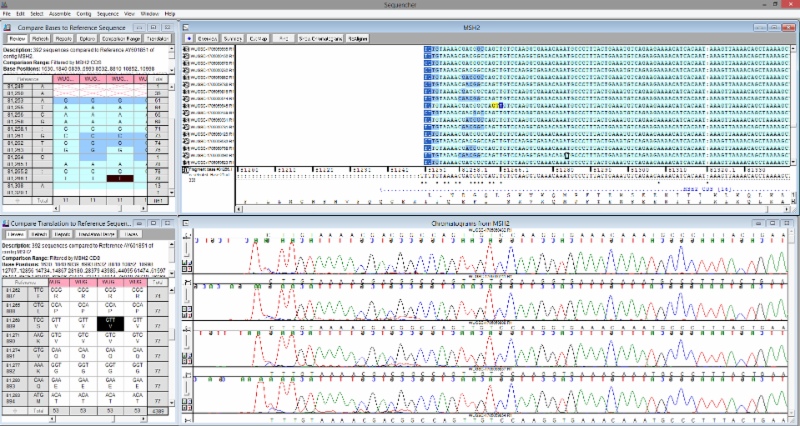
Single Nucleotide Polymorphisms are automatically called and clearly displayed in the table, but what about a heterozygote? Sequencher brings in yet another proprietary feature, Calling Secondary Peaks. This feature allows you to set your secondary trace call threshold, what happens to the base call, and automatically marks the heterozygous bases in the contig. Instead of combing through each trace peak, you can click one button and have all of the heterozygous bases found and flagged.
Sequencher’s feature list doesn’t end with Sanger data. Next Gen data is equally easy to analyze with Sequencher thanks to its host of integrated DNA-Seq Tools. What would analyzing colon cancer data look like using our latest NGS features? It would look a lot like this:
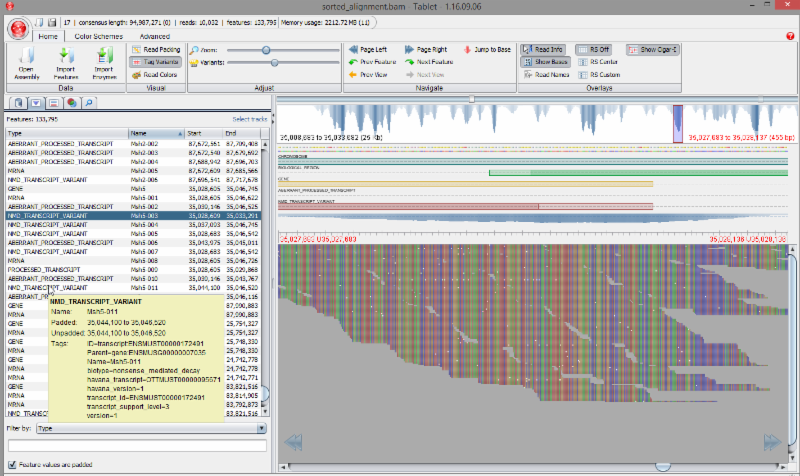
Sequencher can just as easily analyze the entire human genome as it can targeted PCR data. Quickly perform a reference guided alignment using BWA-MEM or GSNAP, and then analyze those alignments for variants using SAMtools Variant Calling. Put it all together in the Tablet NGS viewer complete with Gene Feature Files and Variant Calling Files to fully annotate your findings.
Sequencher doesn’t stop with DNA either. Sequencher has a full RNA-Seq pipeline featuring the Cufflinks suite of algorithms and custom visualization tools waiting at the finish line. No matter what kind of data you’re using, Sequencher has your answer. We continually develop and improve Sequencher based on feedback from our clients worldwide.
With your Pitt email you can access Sequencher for free here!
(content from a 5/2017 Gene Codes Newsletter)